Aktuelle Forschungsauszüge
27. März, 2023 2023-04-13 1:33Aktuelle Forschungsauszüge
Analyse von genomischer Variation
Jede unserer Körperzellen trägt eine vollständige Kopie unserer DNA in sich und damit unser vollständiges Erbgut, das auch als Genom bezeichnet wird. Das Genom ist in Chromosomen organisiert: Wir erben jeweils 23 Chromosomen von unserer Mutter und unserem Vater. Ein Chromosom besteht aus einem sehr langen DNA-Molekül, welches durch spezielle Proteine stabilisiert und räumlich angeordnet wird. Die DNA kodiert genetische Informationen als Abfolge von vier verschiedenen Basen: Adenin (A), Cytosin (C), Guanin (G) und Thymin (T). In diesem Sinne kann ein Chromosom vereinfacht als eine Sequenz der Buchstaben A, C, G und T betrachtet werden. Die Genomforschung beschäftigt sich demnach mit der Frage, wie unsere Genomsequenz individuelle Merkmale beeinflusst, also welche kausalen Zusammenhänge zwischen genetischer Variation und der Ausprägung verschiedener Merkmale bestehen. Viele verschiedene Eigenschaften kön- nen dabei von Interesse sein: Körpergröße, die Anfälligkeit für eine bestimmte Krankheit oder die Intoleranz gegenüber einem bestimmten Arzneimittel.
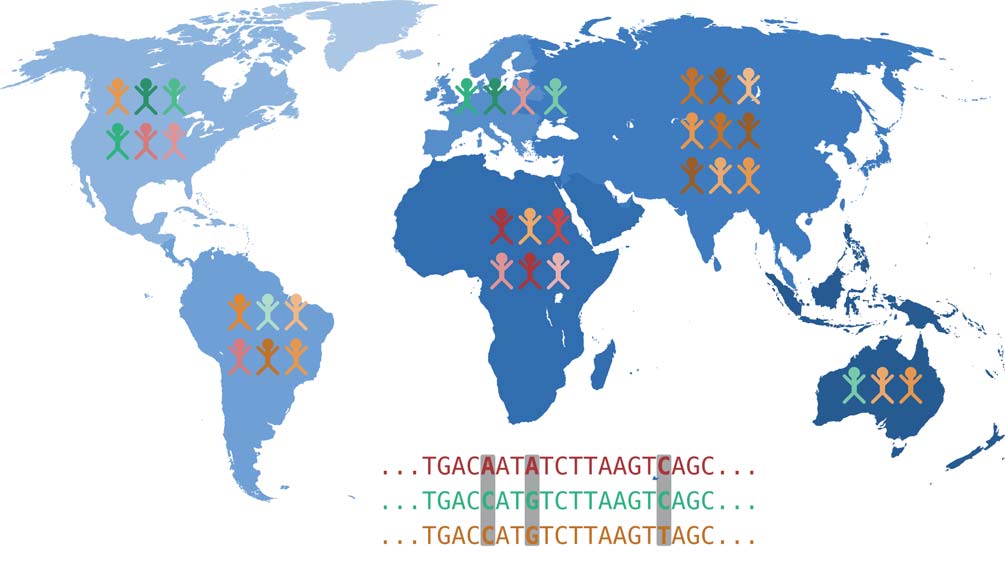
Genetische Veränderungen, die Ursache bestimmter Merkmale sind, befinden sich meistens in den Genen. Dies sind DNA-Regionen, die als Blaupause für Proteine dienen – oder sie liegen in Regionen, die steuern, wie viel von einem ge- wissen Protein produziert wird. Aus den in der DNA kodierten Genen, die man sich wie oben erwähnt als Baupläne vorstellen kann, werden in zwei Schritten Proteine hergestellt. Als erstes werden Kopien dieser DNA-Abschnitte erstellt, die dann als RNA-Moleküle vorliegen. Dieser Vorgang heißt Transkription. Danach werden aus den RNA-Molekülen Proteine produziert, die eine Vielzahl von Funktionen ausüben. Somit wirken sich Änderungen in der DNA über den Umweg der RNA auf die hergestellten Proteine aus. Diese Veränderungen können beträchtliche Auswirkungen auf die Ausprägung von Merkmalen haben und zum Beispiel die Ursache einer genetischen Krankheit sein.
Aber wie können Unterschiede in der DNA oder RNA gemessen werden? In beiden Fällen können hochmoderne Sequenzierverfahren (engl. next generation sequencing, NGS), die ständig weiterentwickelt werden, Verwendung finden. Die Analyse der bei der Sequenzierung entstehenden riesigen Datenmengen stellt Wissenschaftler vor Herausforderungen im Bereich der Informatik, Mathematik und Statistik. Forscher am Zentrum für Bioinformatik stellen sich diesen Herausforderungen und haben wesentlich zur Weiterentwicklung moderner Analysemethoden von DNA- sowie RNA-Sequenzierdaten beigetragen.
Statistisches Lernen in der Bioinformatik
In vielen Bereichen des Lebens ist es in den letzten Jahren zu einem exponentiellen Wachstum von Daten gekommen. Dies liegt zum einen an immer leistungsfähigeren Computern, die in der Lage sind, große Datenmengen zu verarbeiten und zu speichern, zum anderen aber auch an neuen Messmetho-den in vielen Bereichen der Wissenschaft, den sogenannten Hochdurchsatzverfahren. Dies sieht man zum Beispiel in der Physik an Messungen, die am CERN durchgeführt werden, aber auch in der Biologie gibt es immer rasanter ansteigende Datenmengen (z. B. Genom-, Proteom- und Epigenomdaten). Diese Datenflut macht es unmöglich, grundlegende Zusammenhänge in den Daten durch eine reine Betrachtung zu erschließen. So möchten wir bei Genomsequenzen zum Beispiel wissen, an welcher Stelle sich die Gene befinden. Kann man durch zusätzliche Experimente herausfinden, wo sich Gene befinden, so kann man mit Hilfe von statistischen Lernverfahren generelle Sequenzeigenschaften von Genen lernen, die dann in einem Vorhersagemodell verwendet werden können. Dies macht es möglich, für weitere Genomsequenzen die Positionen der Gene vorherzusagen. Diese Vorgehensweise ist insbesondere dann interessant, wenn die zusätzlichen Experimente zeitaufwendig und teuer sind, was für viele Bereiche der Biologie zutrifft.
Die Methoden, die in diesem Szenario eingesetzt werden, gehören zu der Gruppe der statistischen Lernverfahren. Sie werden aber auch in anderen Bereichen angewendet und weiterentwickelt, wie zum Beispiel zum Herausfiltern von Spam Emails, zum Vorhersagen der passendsten Werbung oder zur Vorhersage von Aktientrends. In der Bioinformatik ist das statistische Lernen in allen wichtigen Bereichen vertreten.

Auch am Zentrum für Bioinformatik nimmt das statistische Lernen einen wichtigen Platz ein. So werden zum Beispiel in der Abteilung von Prof. Lengauer schon seit mehr als einem Jahrzehnt neue Verfahren entwickelt, um die Resistenz des HI-Virus gegen bestimmte antivirale Medikamente vorherzusagen. Die Server, die diese Methoden frei zur Verfügung stellen, werden weltweit eingesetzt.
Ein weiterer wichtiger Bereich der Biologie ist die Untersuchung von evolutionären Beziehungen zwischen verschiedenen Spezies. Hierbei werden Stammbäume, die auch phylogenetische Bäume genannt werden, bestimmt. Dort gibt die Entfernung im Baum an, wie weit Organismen evolutionär voneinander entfernt sind. Am ZBI wurden Methoden entwickelt, die das Lernen dieser phylogenetischen Bäume signifikant verbessern.
Ebenfalls von hoher Bedeutung ist der Bereich der Krebsdatenanalyse. Hier ist es interessant, bestimmte Untergruppen von Patienten mit Methoden des unüberwachten Lernens zu finden, aber auch herauszufinden, welche Veränderungen in der Regulation der Proteine einen Einfluss auf den Schweregrad der Krebserkrankung haben. In diesem Bereich hat das ZBI neue Methoden entwickelt, die es zum Beispiel ermöglichen, Messungen verschiedener biologischer Eigenschaften zu vereinen, um eine einfach verständliche Repräsentation der Daten zu erzeugen und darauf basierende Gruppierungen zu finden. Zudem haben wir uns auch mit Fällen beschäftigt, in denen die Messungen für verschiedene Patienten nicht ganz vergleichbar sind und Lernverfahren entwickelt, die diesen Effekt ausgleichen können.
Biologische Netzwerke
Der Stoffwechsel aller Lebewesen basiert auf komplexen biochemischen Interaktionen und Reaktionen hunderttausender von Molekülen, darunter Proteine, DNA- und RNA-Moleküle, Fette, und Zuckermoleküle. Eine zentrale Rolle als Informationsträger spielen die Genome und hier vor allem gewisse Abschnitte in den Genomen, die sogenannten Gene, welche die Information für die Synthese von Proteinen und nicht- Protein-kodierenden RNA-Molekülen speichern.
Dank der rasanten wissenschaftlichen Fortschritte in der Biotechnologie und der Bioinformatik hat sich in den letzten Jahrzehnten unser Wissen über Stoffwechselprozesse so vermehrt, dass allein die Aufbereitung und Speicherung dieses Wissens eine große Herausforderung darstellt: Sie ist nur noch mittels leistungsfähiger bioinformatischer Datenbanken zu bewältigen. Die entsprechenden Daten bilden auch die Grundlage für Simulationen der Stoffwechselprozesse, welche nicht nur neue Erkenntnisse über natürliche biologische Prozesse, sondern auch über die Ursachen von Krankheiten und über pathogene Mechanismen liefern. Diese Erkenntnisse können wiederum zu neuen diagnostischen und therapeutischen Ansätzen führen. Bei entsprechenden Simulationen modelliert man die Stoffwechselprozesse meist mittels Graphen, die auch als biologische Netzwerke bezeichnet werden.
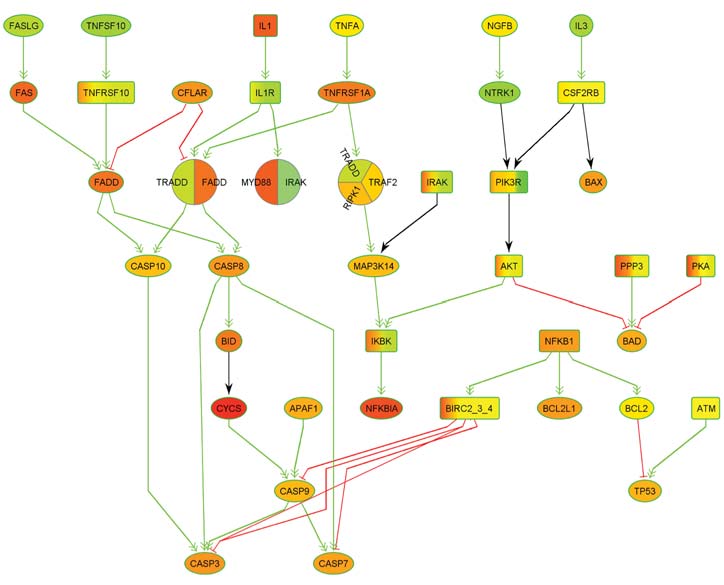
Hierbei repräsentieren die Knoten der Netzwerke die beteiligten Moleküle und die Kanten die biochemischen Interaktionen zwischen den Molekülen. Bei genetisch bedingten Erkrankungen verursachen die vorliegenden genetischen Aberrationen Aktivitätsänderungen in Netzwerken, die zu Störungen in der molekularen Informationsübertragung führen. Die modifizierten Komponenten bezeichnet man auch als deregulierte Prozesse oder Netzwerke. Ein Schwerpunkt der Forschung des ZBI ist die Entwicklung neuer Verfahren für die Detektion deregulierter Prozesse und Subnetzwerke.
Biomoleculare Interaktionen
Biomolekulare Interaktionen sind die Grundlage aller physikalischen Prozesse in lebenden Organismen. In der Tat basieren nahezu alle biochemischen Reaktionen in lebenden Zellen auf molekularen Interaktionen zwischen Proteinen, Nukleinsäuren, Zuckern, Lipiden (Fetten) und niedermolekularen Liganden. Das Verständnis dieser Interaktionen ist eine unabdingbare Grundlage, um die Entwicklung von Organismen und Krankheiten, beziehungsweise die Anfälligkeit für Krankheitskeime nachvollziehen zu können. Mithilfe der Bioinformatik kann man detaillierte Strukturmodelle von molekularen Komplexen erzeugen, alternative Positionen von Wirkstoffen in der Bindungstasche eines Proteins energetisch bewerten, Bindungsprozesse modellieren und darüber hinaus einen Einblick in die theoretischen Grundlagen solcher Interaktionen liefern.
Am Zentrum für Bioinformatik werden Methoden zur Untersuchung von biomolekularen Interaktionen entwickelt.

Epigenetik
Die Epigenetik ist ein Forschungsgebiet, das heute im Zentrum der weltweiten Aufmerksamkeit liegt. Der Grund hierfür ist, dass nach der Sequenzierung des menschlichen Genoms vor zwölf Jahren schnell klar wurde, dass wesentliche Aspekte der Zellregulation nicht direkt aus der genomischen DNA abzulesen sind – denn diese ist ja in allen Zellen gleich. Dagegen unterscheiden sich verschiedene Zellen erheblich voneinander – etwa Zellen in verschiedenen Geweben sowie kranke von gesunden Zellen. Ferner beeinflussen zentrale biologische Prozesse, wie die Antwort auf Stress und das Altern, den Zellzustand umfassend. Diese Faktoren der Zellregulation gehen auf die dynamische Organisation des Genoms im Zellkern zurück, die wiederum auf chemischen Modifikationen der genomischen DNA und des sie umgebenden Molekülgerüsts aus Proteinen beruht.
Die Epigenetik wurde in den 1980er Jahren zunächst als die Wissenschaft der Vererbung solcher Eigenschaften definiert, die sich nicht durch die in DNA codierte Information manifestieren. Es stellte sich dann jedoch schnell heraus, dass solche Vererbungsvorgänge, wie auch die Zellregulation allgemein, auf die komplexe Art gestützt sind, in der das Genom im Zellkern verpackt wird. An dieser Verpackung sind spezielle Proteine beteiligt, die zusammen mit der genomischen DNA das sogenannte Chromatin bilden. Das Chromatin erfüllt zwei Aufgaben. Zum einen muss die genomische DNA, die in ihrer gesamten Länge 2 m misst, in dem Zellkern mit einem Radius von einem tausendstel Millimeter untergebracht werden. Zum anderen sollen die Teile der genomischen DNA, in denen Information codiert ist, die nicht von der Zelle gebraucht wird, vor dem Ablesen geschützt werden. Da die genomische In- formation, die die Zelle benötigt, von dem Gewebe und dem Zellzustand abhängt, ist das Chromatin hoch dynamisch. Seine Struktur wird durch vielfältige chemische Modifikationen der DNA selbst und der sie umgebenden Proteine gesteuert. Die Gesamtheit dieser Modifikationen bezeichnet man als das Epigenom.
Mit in den letzten zwei Jahrzehnten entwickelten molekularbiologischen Techniken können diese Modifikationen (epi-) genomweit vermessen werden. Damit ist das Epigenom kartierbar geworden. Während ein Mensch nur ein Genom hat, hat er im Gegensatz dazu viele Epigenome. Im Prinzip unterscheiden sich die Epigenome in allen der etwa 200 Gewebetypen des Organismus. Darüber hinaus unterscheiden sie sich in jedem Zelltyp in gesunden und krankhaft veränderten Zellen. Die Kenntnis des Epigenoms gilt als Voraussetzung für das Verständnis der Prozesse in dem betre enden Zelltyp. Aus diesem Grund wurde vor einigen Jahren das International Human Epigenome Consortium (IHEC) gegründet – ein weltweiter Zusammenschluss von Wissenschaftlern mit dem Ziel, in den nächsten Jahren mindestens 1.000 Epigenome von menschlichen Zellen zu kartieren. Das Zentrum für Bioinformatik ist an zwei Stellen an IHEC beteiligt (siehe Projekte DEEP und BLUEPRINT).
Infektionskrankheiten
Infektionskrankheiten sind für die medizinische Forschung eine ständige Herausforderung. Der Grund liegt darin, dass sich in der Natur seit frühester Zeit ein harter Wettbewerb zwischen den verschiedenen Spezies entwickelt hat. Dieser Wettbewerb ist auch heute noch in vollem Gange. Uns Menschen wird er besonders durch die Aktivität von krankheitserzeugenden Erregern bewusst. Der Erreger sucht seinen Vorteil im Infizieren oder Besiedeln des menschlichen Wirts. Der infizierte menschliche Organismus wehrt sich gegen den Erreger mithilfe seines Immunsystems und, in neueren Zeiten, unterstützt durch eine medikamentöse Therapie. Der Erreger wiederum reagiert darauf durch eine Anpassung an den Wirt. Diese Anpassung findet nicht vornehmlich im einzelnen Erreger statt, sondern vielmehr durch eine evolutive Veränderung des Genoms des Erregers in den folgenden Generationen. Sie bringen damit Eigenschaften hervor, die zukünftig für das Überleben im Wirt günstig sind. Durch die ständige Veränderung des Erregers müssen dann immer wieder neue therapeutische Strategien entwickelt werden – es findet ein Wettrennen zwischen dem Erreger und uns Menschen als Wirten statt.
Aufgrund ihrer hohen Relevanz für die menschliche Gesundheit aber auch aufgrund des beispielhaften evolutiven Szenarios, das ihnen zu Grunde liegt, stellen Infektionskrankheiten einen reichen Fundus an hoch interessanten und relevanten Forschungsproblemen dar. Das Zentrum für Bioinformatik hat dieses Gebiet zu einem seiner Schwerpunkte gewählt. Wir untersuchen Infektionskrankheiten aus einer Vielzahl von Perspektiven. Dabei steht allerdings das evolutive Wettrennen zwischen Wirt und Erreger im Vordergrund.
Im Zentrum untersuchen wir Infektionskrankheiten mit besonders hoher evolutiver Dynamik. Die höchste Dynamik hat dabei HIV vorzuweisen, das eine hohe Mutationsrate besitzt und sich daher rasant im Körper eines jeden Patienten weiter entwickeln kann. Im Zentrum wird HIV auf verschiedenen Ebenen beforscht.

Im Vergleich zu Viren haben Bakterien ein wesentlich komplexeres Genom. Das ermöglicht ihnen auf vielfältigere Art, sich an einen Wirt oder an eine medikamentöse Therapie anzupassen. Aus diesem Grund folgt der Fortschritt im Bereich der Forschung über bakterielle Infektionen dem über virale Infektionen. Die Resistenzentwicklung bei Bakterien ist dabei jedem im Zusammenhang mit der zurückgehenden Effektivität von Antibiotika bewusst. Im Zentrum untersuchen wir solche Resistenzphänomene und nutzen bakterielle Eigenschaften aus, um neue Medikamente zu entwickeln. Schließlich forscht das Zentrum an einem groß angelegten Projekt, das das Ziel hat, den bereits erfolgreichen Zugang zur bioinformatischen Analyse von viraler Resistenz auf Bakterien zu übertragen.
Krebs
Trotz der beachtlichen Fortschritte der Onkologie in den letzten Jahrzehnten ist Krebs auch heute noch für 13% aller Todesfälle weltweit verantwortlich und damit direkt nach kardiovaskulären Erkrankungen eine der Haupttodesursachen.
Die Ursachen von Krebs sind multifaktoriell. Sowohl genetische Aberrationen als auch Umwelteinflüsse und Lebensstil tragen zur Entstehung von Krebs bei. Heute beruht die Behandlung immer noch weitgehend auf Operation, Bestrahlung und Chemotherapie. Obwohl die meisten Patienten auf diese Behandlungen ansprechen, werden nicht alle geheilt. Ein Hauptgrund hierfür ist die Heterogenität von Tumoren, die vor allem auf eine in Tumoren ablaufende „zelluläre Evolution” zurückzuführen ist. Diese wird wiederum durch hohe Zellteilungs- und Mutationsraten und ein Versagen der zellulären Reparaturmechanismen ausgelöst. Vereinfacht dargestellt bedeutet dies, dass ein Tumor sich in der Regel aus genetisch unterschiedlichen Tumorzellen zusammensetzt. Viele dieser Zellen können durch eine Therapie getötet werden, andere Tumorzellen wiederum sind entweder bereits resistent gegen die Therapie oder aus ihnen können sich durch das Auftreten neuer Mutationen resistente Zellen bilden.
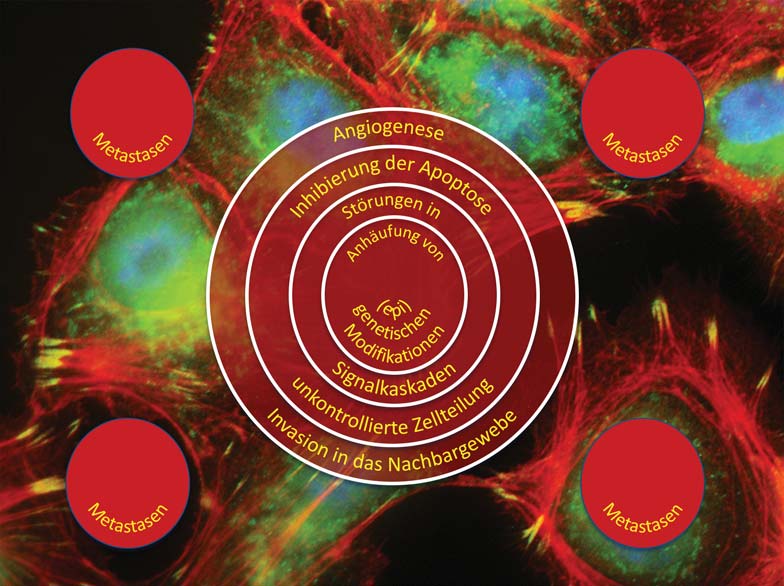
Die Heilungschancen bei vielen Krebsarten ist immer noch stark davon abhängig, in welchem Entwicklungsstadium die Erkrankung diagnostiziert wird. Hier gilt immer noch die einfache Aussage: Je früher, desto besser. Bei der Früherkennung spielen neben bildgebenden Verfahren nicht-invasive, auf Biomarkern in Körperflüssigkeiten basierende Diagnoseansätze eine zentrale Rolle. Arbeitsgruppen des ZBI haben für eine Reihe von Krebserkrankungen neue, auf hochdimensionalen Profilen von Tumorantigenen und MicroRNAs basierende, diagnostische Ansätze entwickelt. Diese besitzen eine höhere Genauigkeit als die zurzeit zugelassenen Diagnoseverfahren. Neben zahlreichen Publikationen sind aus diesen Projekten auch eine Reihe von Patenten hervorgegangen. Für ein Verfahren werden bereits die für eine Kommerzialisierung notwendigen Validierungsstudien durchgeführt. Sie zeigen bis dato sehr vielversprechende Ergebnisse.