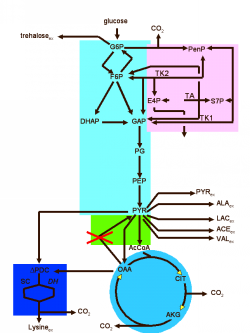
Forschungsthemen im Bereich der Bioinformatik
27. März, 2023 2023-04-13 1:33Forschungsthemen im Bereich der Bioinformatik

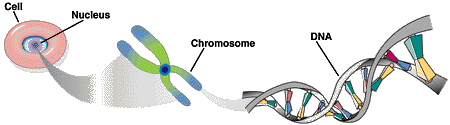
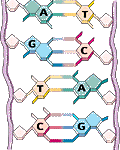
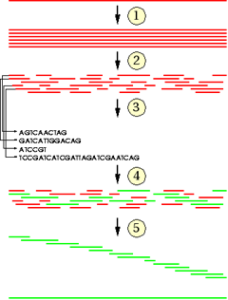
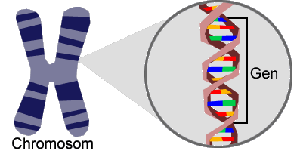
Jedes Chromosom besteht aus einem DNA-Doppelstrang: Zwei Einzelstränge winden sich in Form einer Doppelhelix umeinander. Jeder DNA-Einzelstrang besteht aus einer Abfolge von Bausteinen (Nukleotiden), die durch die jeweils enthaltene Base charakterisiert sind.
Es gibt insgesamt vier verschiedene Bausteine, die mit den vier Buchstaben A, C, G und T bezeichnet werden. Je zwei Nukleotide (A+T, G+C) sind komplementär, das heißt sie binden aneinander. Auf diese Weise bildet sich der Doppelstrang. Von den beiden Einzelsträngen wird daher nur einer benötigt, da der jeweils andere aufgrund der Basenkomplementarität eindeutig definiert ist. Das menschliche Genom kann daher als lange Zeichenkette bestehend aus den vier Buchstaben A, T, G und C dargestellt werden.