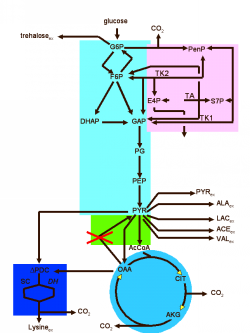
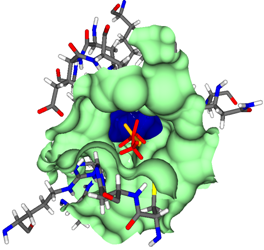
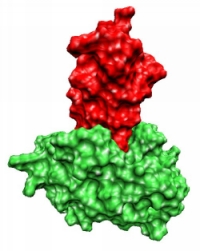
Research Topics in the Field of Bioinformatics
March 27, 2023 2023-04-13 1:33Research Topics in the Field of Bioinformatics

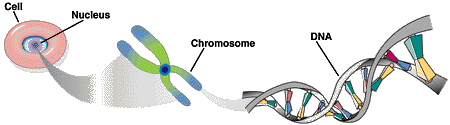
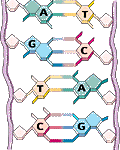
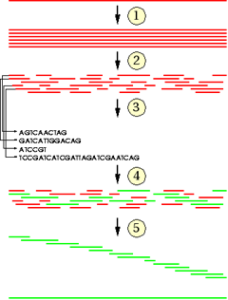
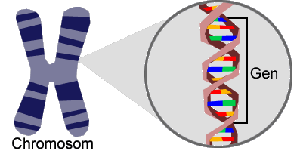
Each DNA molecule consists of two strand in the form of a double helix. Each DNA strand is a linear polymer that consists of similar subunits (monomers) connected end to end.
Within each monomer one can find a sugar, a phosphate and a base component. The sequences of bases represents a form of linear infomation. There are four bases denoted by the letters A, C, G and T. The bases A,T and G,C are complementary, i.e. bind to each other. Based on this base pair complementarity a single strand contains the full genetic information.